
Basics
To run Gene Stacker, unzip the downloaded package genestacker-[VERSION].zip, open a terminal window (command prompt on Windows) and navigate
to the unzipped folder genestacker-[VERSION]:
$ cd [...]/genestacker-[VERSION]
where [...] should be replaced with the path of the directory in which genestacker-[VERSION].zip
has been unzipped and [VERSION] should be replaced with the appropriate version number.
To check if you are ready to run Gene Stacker, type
$ java -jar genestacker.jar -help
which should print brief usage information including an overview of all options.
If not, you probably don't have a (recent) Java Runtime Environment installed. In this case,
first download and install the latest JRE from
this page and then retry
to run Gene Stacker.
Now you are ready to use Gene Stacker on one of the example input files. There are two required parameters:
-g,--max-gen <g>, which sets the maximum number of generations of the constructed crossing schedules,
and -s,--success-prob <p>, which specifies the desired total success rate (controlling the
computed population sizes). For example, type
$ java -jar genestacker.jar -g 3 -s 0.95 examples/A.xml out
or (long version)
$ java -jar genestacker.jar --max-gen 3 --success-prob 0.95 examples/A.xml out
to run Gene Stacker on example input A, with a maximum of 3 generations and a total success rate of 0.95. Note that on Windows,
you can also use backslashes as file separators in the input and output file paths. Output will be stored in a zip file
out.zip containing several files for each constructed schedule
as explained in the output section below.
The input files of Gene Stacker are XML files with a custom structure, see the input section for more information and instructions on how to create your own input files. Finally, Gene Stacker provides a lot of options including a wide range of heuristics and constraints, which are discussed in the next section. Note that specifying additional constraints and/or using other heuristic presets might significantly influence the execution time.
Options
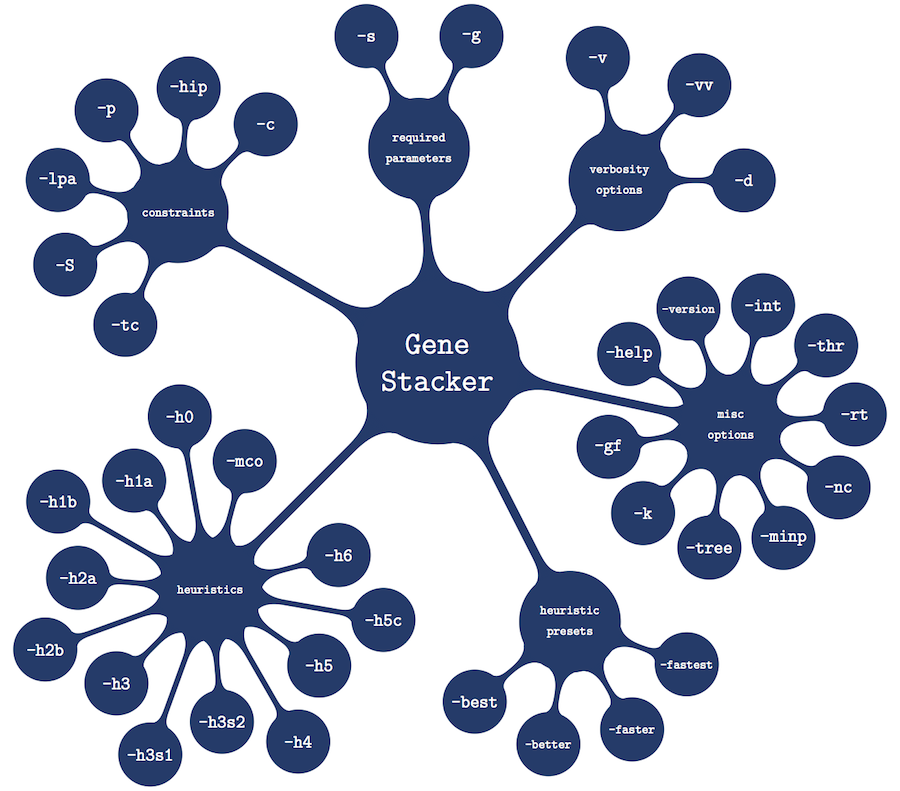
The figure below shows a graphical visualization of all options available in Gene Stacker. Click on any option or option group for more details. All options have a short version, starting with a single hyphen, and a long version, starting with a double hyphen. Short and long versions are separated with a comma sign in the descriptions and can be used interchangeably. For some options, the short and long version are identical. Furthermore, note that options are case sensitive. To get an overview of all options on the command line, run
$ java -jar genestacker.jar -help
R Interface
To use the included R interface, open the R console and change your working directory to the unzipped Gene Stacker folder, for example:
> setwd("[...]/genestacker-[VERSION]")
where [...] should be replaced with the directory in which the downloaded Gene Stacker package has been unzipped, and
[VERSION] should be replaced with the appropriate version number. For example, on Unix, the full command could be
> setwd("~/Documents/genestacker-v1.7")
or something like this on Windows:
> setwd("C:/Users/John/Documents/genestacker-v1.7")
Then, load the Gene Stacker script using
> source("genestacker.R")
To check whether Gene Stacker has been successfully loaded into R, try printing its version number by running
> genestacker.version() Gene Stacker v1.7If this works, you are ready to run Gene Stacker from R using the function
genestacker.run with the following general interface:
> genestacker.run(input, output, params, ...)
The parameter input refers to the input file and output indicates the output file path. These are
followed by a sequence of parameters param=... including at least the maximum number of generations g
and desired overall success rate s. For example:
> genestacker.run("examples/A.xml", "out", g=3, s=0.95)
For almost all CLI options described above,
the R interface provides a corresponding parameter named after the short version of the CLI option, without the hyphen.
Boolean flags, i.e. options that do not require a parameter
value, can be switched on by setting param=TRUE, e.g. v=TRUE. For options that do require
a parameter value val, use param=val, e.g. c=4. The following example specifies some
additional constraints and uses the preset faster:
> genestacker.run("examples/A.xml", "out", g=3, s=0.95, c=4, p=5000, S=300, preset="faster")
Input
Gene Stacker reads its input from custom XML files, structured according to the Gene Stacker input XML schema. An example is given by
<?xml version="1.0" encoding="UTF-8" standalone="no"?>
<genestacker_input xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xsi:noNamespaceSchemaLocation="http://genestacker.ugent.be/xml/genestacker_input.xsd">
<initial_plants>
<plant>
<genotype>
<chromosome>
<haplotype targets="0"/>
<haplotype targets="0"/>
</chromosome>
<chromosome>
<haplotype targets="101"/>
<haplotype targets="010"/>
</chromosome>
</genotype>
</plant>
<plant>
...
</plant>
</initial_plants>
<ideotype>
<genotype>
<chromosome>
<haplotype targets="1"/>
<haplotype targets="1"/>
</chromosome>
<chromosome>
<haplotype targets="111"/>
<haplotype targets="101"/>
</chromosome>
</genotype>
</ideotype>
<genetic_map>
<distances_on_chromosome/>
<distances_on_chromosome>
<dist cM="31"/>
<dist cM="42"/>
</distances_on_chromosome>
</genetic_map>
</genestacker_input>
which corresponds to the example input A.xml that is included in the downloadable Gene Stacker package. First, the initial
parental genotypes are described inside the <initial_plants> tag. For each genotype, an equally long series of chromosomes is listed.
Each chromosome consists of two haplotypes, which contain an equal number of binary target alleles. Subsequently, the genotype of the ideotype
is specified within the <ideotype> tag,
followed by the genetic map within the <genetic_map> tag.
This map contains the distances between subsequent loci on the same chromosome, in
centimorgan units (cM). For chromosomes with a single target locus, an empty <distances_on_chromosome/> tag
is included. For any chromosome with \(k \ge 2\) target loci, \(k-1\) distances are specified using a series of
<dist cM="..."/> tags, inside the <distances_on_chromosome> tag
corresponding to the respective chromosome.
To create your own input files, follow the structure from the example files and the explanation given above. It is possible to validate your input files by including the attributes
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xsi:noNamespaceSchemaLocation="http://genestacker.ugent.be/xml/genestacker_input.xsd"in the
<genestacker_input> root tag, as shown in the example above, using an XML schema validator such as the online
tool from Core Filing. Upload your input file
there as XML instance, and press validate to check whether it contains valid Gene Stacker input. Gene Stacker also validates all input
files loaded for execution and informs the user on any issues with the input format at runtime.
Output
Gene Stacker creates a zip file that contains the following three files for each constructed crossing schedule:
-
A visualization
scheme[i].pdf, created using the Graphviz software. By default the pdf format is used, but this can be changed using the option-gf,--graph-file-format <f>(see the options section). If Gene Stacker has not been correctly linked with Graphviz, as explained in the installation section of the download page, no such visualizations will be produced. -
A graphviz file
scheme[i].graphviz, which can be used to manually generate visualizations, for example by executing$ dot -Tpdf -o scheme1.pdf scheme1.graphvizwhich will create a pdf visualization of the crossing schedule described inscheme1.graphviz. Note that one Windows machines,dotshould be replaced with the full path to thedot.exeexecutable, see the installation section for more details on how to find out this path. Alternatively, you can use an online Graphviz viewer such as GraphViz Workspace to generate figures, by opening thescheme1.graphvizfile with your favorite text editor and copy pasting the contents into GraphViz Workspace. -
A custom XML file containing a structured representation of the crossing schedule, following the
Gene Stacker crossing schedule XML schema. This
file can be useful in case you want to perform further automated experiments or simulations with the
output of Gene Stacker, as the complete structure and all important features of the schedule can easily
be parsed from this file. An example is given below.
<?xml version="1.0" encoding="UTF-8" standalone="no"?> <crossing_scheme xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" gamma="0.9" gammaPrime="0.9486832980505138" numGen="3" lpa="0.0" totalPopSize="2414" xsi:noNamespaceSchemaLocation="http://genestacker.ugent.be/xml/crossingscheme.xsd"> <seedlots> <seedlot generation="2" id="s4x0"> <used_seeds amount="2399" generation="2"/> </seedlot> <seedlot generation="0" id="s0x0"> <used_seeds amount="1" generation="0"/> <used_seeds amount="1" generation="1"/> </seedlot> ... </seedlots> <plants> <plant generation="1" id="p4x0" obsProb="1.0" lpa="0.0" seedlot="s2x0"> <genotype> <chromosome> <haplotype targets="0001"/> <haplotype targets="1110"/> </chromosome> </genotype> </plant> ... </plants> <crossings> <selfing id="c17" plant="p21x0" seedlot="s11x0"/> <crossing id="c9" plant1="p1x0" plant2="p0x1" seedlot="s2x0"/> <crossing id="c8" plant1="p4x0" plant2="p0x0" seedlot="s4x0"/> </crossings> </crossing_scheme>The XML output specifies the overall success rate (gamma), derived success rate per targeted genotype (gammaPrime), number of generations (numGen), overall linkage phase ambiguity (lpa) and total population size (totalPopSize) as attributes of the<crossing_scheme>root tag.Subsequently, all seed lots are listed, indicating the generation in which they have been created through crossings and the number of seeds taken from each seed lot in each generation. Below, the targeted plants are stated, specifying from which seed lot they are grown, and in which generation. Also, the probability of observing the target genotype is stated (
obsProb), as well as the corresponding linkage phase ambiguity (lpa). Finally, the performed crossings and selfings are listed, indicating which plants are crossed and which seed lot is obtained from the crossing. Crossings are always part of the same generation as the plants that are crossed and lead to a seed lot at the beginning of the next generation.